Imagens

História clínica (com exames complementares pertinentes):
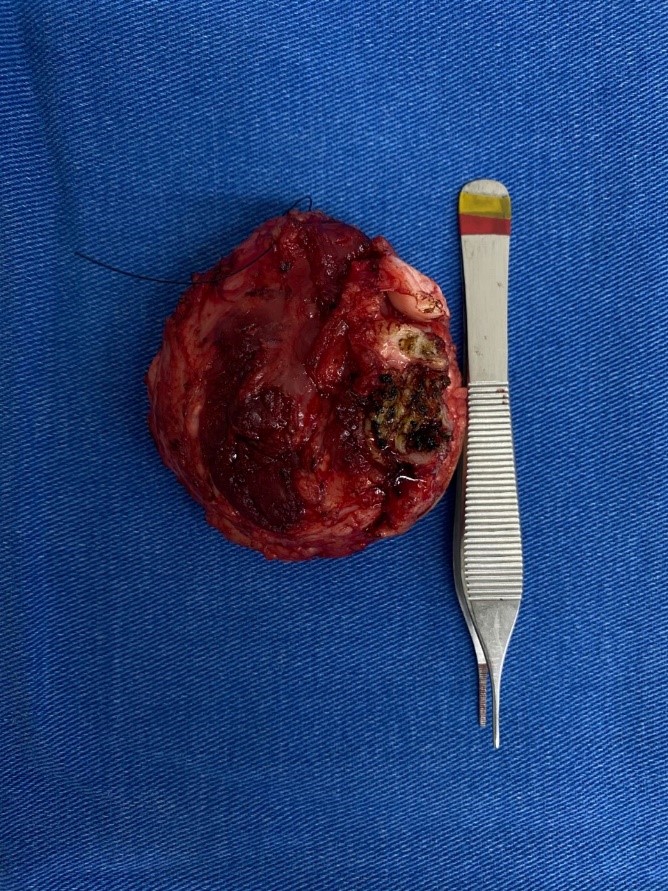
Paciente G.S.R., sexo masculino, 2 anos de idade, apresenta formação expansiva em fossa poplítea de perna esquerda. O exame de imagem por Ressonância magnética de perna e joelho esquerdo evidenciou formação expansiva na fossa poplítea com epicentro nos ventres musculares / espaços intermusculares do compartimento póstero-lateral do terço proximal da perna. A lesão media 8,0 x 4,5 x 6,1 cm ( CC X AP X LL) envolvendo a porção proximal do feixe vásculo-nervoso poplíteo. O paciente foi encaminhado para realização de biópsia com a hipótese diagnóstica de Fibrossarcoma infantil.
Imagens



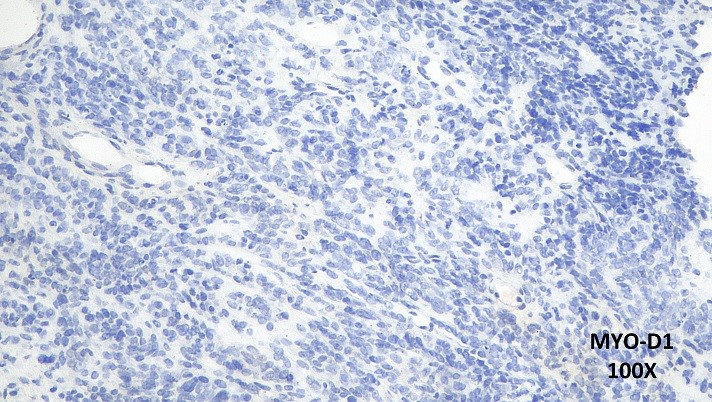
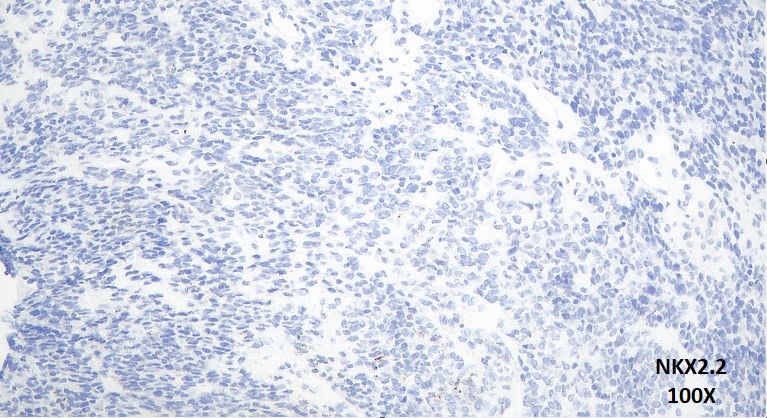
Haundout (achados anatomopatológicos, diagnóstico diferencial, seguimento clínico, discussão):
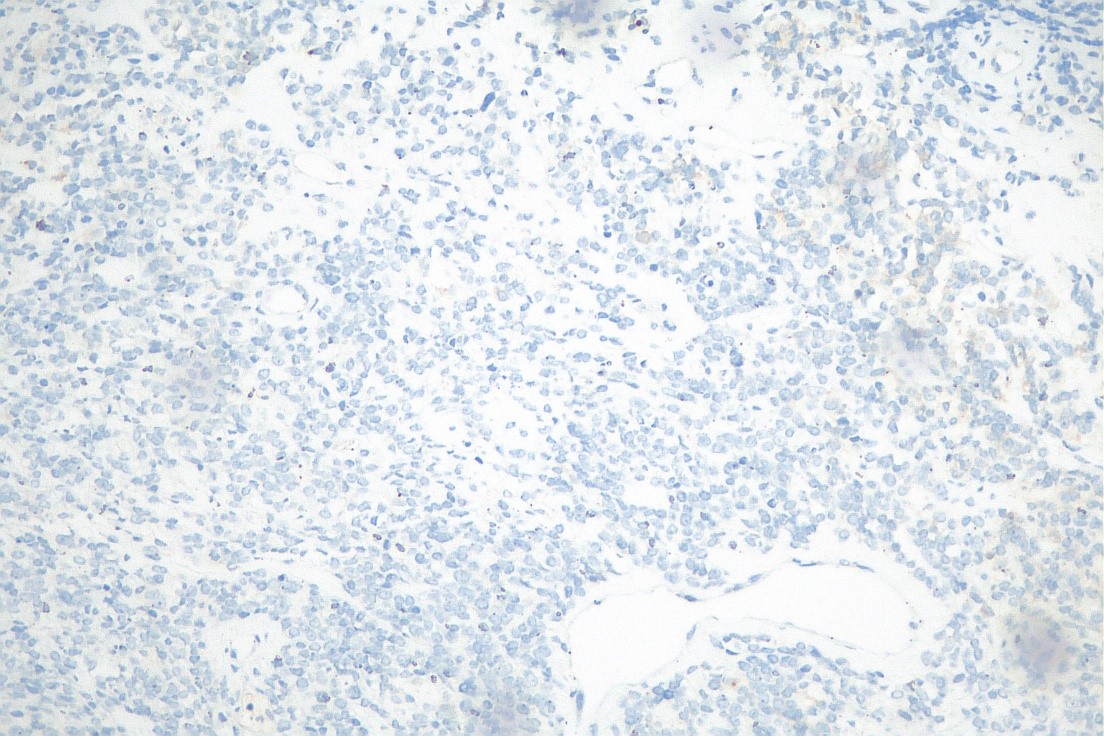
Os cortes histológicos da biopsia por agulha demonstraram proliferação de células redondas e azuis com área focal de células alongada com núcleos brandos.
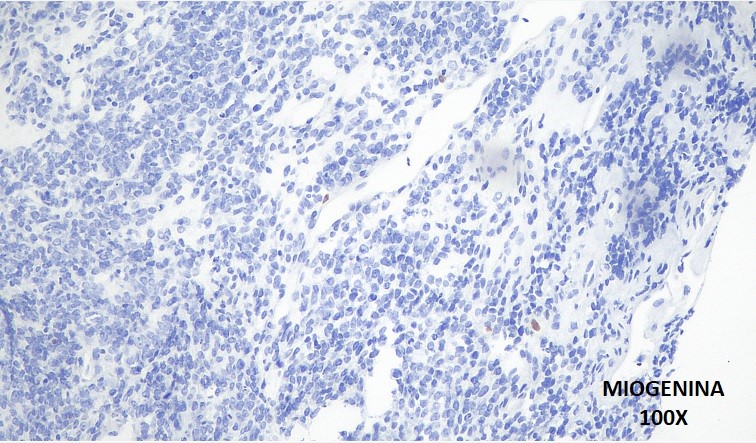
O estudo imuno-histoquímico demonstrou positividade focal para CD99, CD56 e SALL4 e negatividade para EMA, Miogenina, Actina de músculo liso, AE1/AE3, CD34, FLI1, NKX2.2, Cromogranina, Sinaptofisina, CD30, CD45, S100, ERG, Mieloperoxidase, SOX-10, GLYPCAN, HMB45, MELAN-A, Alpha-1 Fetoproteína, Neurofilamento, WT-1, D2-40, Desmina, MYO-D1 e OCT-4. O índice proliferativo pelo imunomarcador ki-67 foi de 50%.
Observou-se ainda perda de imunoexpressão para INI1 como imunomarcação negativa para pan-TRK corroborada pelo painel de fusão gênica – Sequenciamento genético (NGS) .
O conjunto de achados morfológicos imuno-histoquímicos favorecem o diagnóstico de Tumor rabdoide extra-renal.
O tumor rabdoide extrarrenal é um tumor de partes moles altamente maligno, acometendo principalmente lactentes e crianças. Morfologicamente é caracterizado por células rabdóides dispostas em lençóis ou em padrão trabecular sólido, contornos arredondados com núcleos em forma de feijão, nucléolos proeminentes e abundante citoplasma. Os tumores rabdóides extra-renais têm uma incidência de deleções homozigóticas do gene SMARCB1 (raramente SMARCB-4) decorrida de translocações cromossômicas. Assim, os tumores rabdóides mostram perda de expressão SMARCB1 que pode ser demonstrada por imuno-histoquímica através da marcação nuclear da proteína INI1. Em nosso caso, observa-se perda da imunoexpressão para INI1 com controle interno de células endoteliais positivas.
O principal diagnóstico diferencial morfológico para presente relato é o Fibrossarcoma infantil (FI). Assim como nos casos de TRER, o FI é uma neoplasia que ocorre na infância em até o primeiro ano de vida. Consiste em um tumor fibroblástico maligno de rápido crescimento, caracterizado por células monomórficas fusiformes a ovóides/arredondadas com núcleos ligeiramente angulados. Frequentemente está relacionado com fatores genéticos envolvendo a fusão ETV6-NTRK3 e ou outros rearranjos envolvendo NTRK1, BRAF e MET. Em nosso caso essas alterações genéticas foram descartadas pela imuno-histoquímica pan-TRK e o sequenciamento genético.
Outros diagnóstico diferenciais são doenças linfoproliferativas, Rabdomiossarcoma e tumores de células germinativas que foram excluídos pelos demais marcadores imuno-histoquímicos .
Após o diagnóstico, o paciente foi encaminhado para tratamento quimioterápico com subsequente ressecção cirúrgica da lesão. O material foi submetido ao exame anatomopatológico para avaliação de resposta terapêutica. Observou-se menos de 5% de neoplasia residual.
Referências bibliográficas:
- Rosai, J., Ackerman, L. V., Goldblum, J. R., Lamps, L. W., McKenney, J. K., & Myers, J. L. (2018). Rosai and Ackerman’s Surgical Pathology. Elsevier.
- World Health Organization. (2020). Soft tissue and bone tumours (5ª).



 Bourbon Resort Cataratas do Iguaçu
Bourbon Resort Cataratas do Iguaçu Av. das Cataratas, 2345 - Vila Yolanda,
Av. das Cataratas, 2345 - Vila Yolanda,